Generation and compilation of data required for regulatory approval
Documentation of the physicochemical properties of drug candidates and formulations was carried out in the format specified by the New Drugs and
Clinical Trial Rules, 2019. Regulatory data on specifications of physico-chemical properties (Chemistry, Manufacturing and Controls) of four
CSIR-CDRI candidate drugs: S007-867, S-007-1500 and S-011-1793 were compiled. Investigational New Drug Applications to the Drugs Controller
General of India for clinical testing of these candidate drugs are being prepared before submission.
Development, validation and deployment of methods of pharmaceutical analysis
Analytical methods were developed and validated according to the New Drugs and Clinical Trials Rules, 2019, for several new as well as known drugs.
This year, pharmaceutical analysis of 23 different kinds of samples of synthetic compounds, plant products and industrial production batches were
analysed; about half the samples analysed in the previous year. Another set of about 1000 samples were analysed for drug content, content uniformity,
drug release, stability and impurity profiling in formulation development activities. There are 14 different active projects on pharmaceutical
analysis in progress currently. The average time from receipt of sample to filing an analytical report this year was 36 days, up from 27.9 days in
the previous year.
Inhalable particles containing anti-tuberculosis agents
The grant application for funds required for a Phase-1 b was reviewed and revised as per expert comments. The clinical testing plan has been submitted
to the Institutional Ethics Committees of CSIR-CDRI and King George’s Medical University. The trial has objectives of determining safety,
pharmacokinetics and early measurement of drug activity.
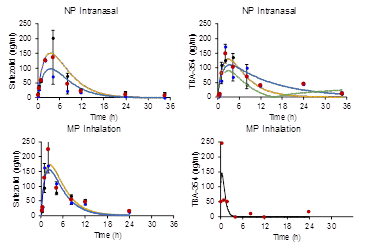
Inhalable particles containing drugs used in multi-drug resistant (MDR) tuberculosis
In collaboration with Prof. Gareth W Griffiths at the University of Oslo, Prof. Andrew Thompson from The University of Auckland, inhalable particles
containing sutezolid, TBA-354 and Compound 32625 were prepared and evaluated against M. tuberculosis strain H37Rv in THP-1 derived macrophages.
Pharmacokinetics following inhalation of micron-sized particles and intranasal instillation of nanoparticles of identical composition were
established in Guinea pigs. Evaluation in a mouse model of tuberculosis is in progress at National JALMA Institute of Leprosy and Other
Mycobacterial Diseases, Agra.
Transient transfection of the respiratory epithelium with gamma interferon to provide host-directed therapy in pulmonary tuberculosis
Pulmonary tuberculosis (PTB) is caused by intracellular pathogens like Mycobacterium tuberculosis (Mtb) and congeners in a fraction of the population
that is infected by the bacteria. Nebulized gamma interferon (IFN-) has shown clinical efficacy against multiple drug resistant (MDR) PTB.
Because IFN- protein is expensive, thermolabile and requires a cold chain for field deployment, it is not suitable for limited resource settings.
We prepared a dry powder for inhalation (DPI) containing DNA constructs polyplexed with poly (ethyleneamine) (PEI) to permit transient transfection
of the lung and airway epithelium with IFN- or with fluorescent proteins to enable imaging. The median diameter of the DPI particles by electron
microscopy was 1.2 µm and the mass median aerodynamic diameter (MMAD) was 2.85±1.8 µm GSD. DNA incorporated in the particles remained intact. A549
cells exposed to particles showed transient expression of green fluorescent protein (GFP) from 6-24 h, after which the protein was observed to
co-localize with lysosomes. Mice receiving inhalations were live imaged for red fluorescent protein and expression of IFN- in bronchio-alveolar
lavage fluid. Both signals peaked at about 24h after inhalation and declined by about 36h. Particles inhaled by Mtb-infected mice bearing
granulomatous lesions induced extensive autophagy. We concluded that the formulation provides controlled release of functional IFN- in a
time-window suitable for investigation of preclinical efficacy and safety as a host-directed therapy of pulmonary TB.
Inhalable D-cycloserine and ethionamide to overcome pharmacokinetic interaction and impart bactericidal efficacy to drugs considered as bacteriostatic agents
Pharmacokinetics and efficacy of a combination of D-cycloserine (DCS) and ethionamide (ETO) via oral and inhalation routes were studied in mice.
The plasma t1/2 of oral ETO at human-equivalent dose reduced from 4.63±0.61 h to 1.64±0.40 h when DCS was co-administered. AUC(0-t) reduced to
one-third. Inhalation overcame the interaction, restoring primary pharmacokinetic parameters to expected values. Inhalation, but not oral doses
reduced lung CFU/g of Mycobacterium tuberculosis H37Rv from 6 to 3 log10 in four weeks, indicating bactericidal activity of inhaled combination;
whereas the two drugs are classified as bacteriostatic agents by the WHO.

Formulations and Targeted Delivery
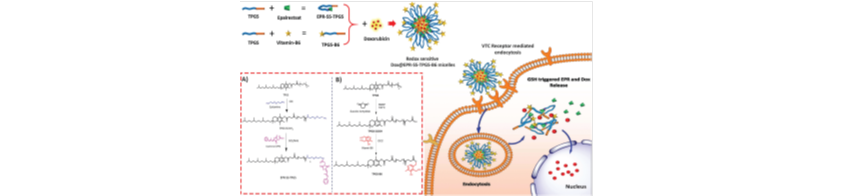
Targeted co-delivery of the aldose reductase inhibitor epalrestat and chemotherapeutic doxorubicin via a redox-sensitive prodrug approach promotes synergistic tumor suppression
Rapidly growing evidence suggests a strong dependence of a polyol pathway enzyme Aldose Reductase (AR) in cancer progression and invasion. Thus,
inhibiting the AR through therapeutic inhibitors has a potential application in cancer treatment. Epalrestat (EPR) is the only marketed AR inhibitor
with proven safety and efficacy in the management of complications like diabetic neuropathy. However, its short half-life and highly hydrophobic
nature restrict its use as an anticancer agent. In the present study, we first developed a redox-sensitive prodrug of EPR by conjugating Tocopherol
Polyethylene Glycol Succinate (TPGS) which can form a self-assembled micellar prodrug (EPR-SS-TPPGS). Subsequently, to achieve synergistic
chemotherapeutic efficacy Doxorubicin (Dox) was co-loaded into the EPR-SS-TPGS micelles where the system is disrupted in a tumor redox environment
and co-delivers Dox and EPR in a ratiometric manner. We then employed TPGS conjugated vitamin-B6 as a targeting moiety and prepared the mixed
micelles to facilitate VTC receptor-mediated uptake. The encapsulation of Dox and EPR with the developed prodrug approach showed significant
synergies with increased intracellular accumulation and redox triggered release in MDA-MB-231 and 4T1 cell lines leading to superior cell cycle
arrest, mitochondrial membrane potential, and apoptosis. Prolonged circulation half-life and tumor site bioavailability were achieved for both
the drugs with the developed approach. Surprisingly, EPR and Dox combination significantly down-regulated the CD44 receptor expression which is
the main contributing factor of tumor metastasis. Furthermore, in vivo evaluation demonstrated a significant reduction in Dox-induced
cardiotoxicity. In summary, this nanoencapsulation paradigm of AR inhibitors with chemotherapeutic agents lays the foundation of new opportunities
in combination chemotherapy. Biomater. Sci., 2019, 7, 2889
Multifunctional hybrid nanoconstructs facilitate intracellular localization of doxorubicin and genistein to enhance apoptotic and antiangiogenic efficacy in breast adenocarcinoma
The progressive development of tumors leading to angiogenesis marks the advancement of cancer which requires specific targeted treatment preferably
with combination chemotherapy. However, there is still a long way to go to develop an efficient delivery system that could overcome the tumor
microenvironment to achieve efficient delivery. Herein, we have developed spermine (SPM) tethered lipo-polymeric hybrid nanoconstructs with two
tier strategies, cell surface heparan sulfate proteoglycans (HSPG) specificity for higher intracellular localization and pH dependent charge reversal
in the tumor microenvironment (below pH 5.8) to facilitate Doxorubicin (Dox) and Genistein (Gen) release in a synergistic combination. We have
observed the specific uptake of SPM anchored hybrid nanoconstructs by receptor-mediated endocytosis in human breast cancer cells (MDA-MB-231) through
the HSPG receptor. The SPM-D + G/NPs induced a higher rate of apoptosis in MDA-MB-231 cells via disruption of the mitochondrial membrane potential
and also exhibited a stronger anti-angiogenic effect governing the inhibition of VEGF pathway modulation, proliferation, invasion and migration of
HUVECs in in vitro and in vivo Balb/c mouse models. The involvement of Akt/Hif1α/VEGF dependent signal cascading and its down-regulation with a
pro-apoptotic drug Dox and an anti-angiogenic agent Gen was evident as demonstrated by an in silico docking study and subsequently proven by
RT-PCR and western blotting. Altogether this study highlights the potential role of SPM in targeting HSPG receptors and synergistic delivery of
Dox and Gen as a promising strategy to effectively inhibit BAC progression and these findings could open a new window to deliver combinations of
chemotherapeutic agents along with anti-angiogenic ligands using hybrid nanoparticles. Biomaterials Science, 2020, DOI: 10.1039/C9BM01246J

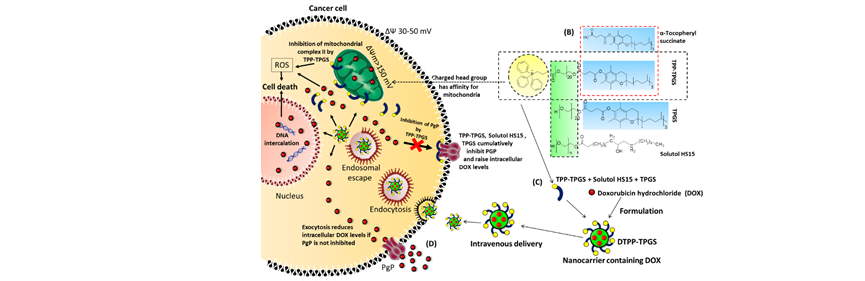
Induction of mitochondrial cell death and reversal of anti-cancer drug resistance mediated via nanocarrier composed of triphenylphosphonium derivative of TPGS
We have devised a nanocarrier using “tocopheryl polyethylene glycol succinate (TPGS) conjugated to triphenylphosphonium cation” (TPP-TPGS) for
improving the efficacy of doxorubicin hydrochloride (DOX). Triphenylphosphonium cation (TPP) has affinity for an elevated transmembrane potential
gradient (mitochondrial), which is usually high in cancer cells. Consequently, when tested in molecular docking and cytotoxicity assays, TPP-TPGS,
owing to its structural similarity to mitochondrially directed anticancer compounds of the “tocopheryl succinate” family, interferes specifically in
mitochondrial CII enzyme activity, increases intracellular oxidative stress, and induces apoptosis in breast cancer cells. DOX loaded nanocarrier
(DTPP-TPGS) constructed using TPP-TPGS was positively charged, spherical in shape, sized below 100 nm, and had its drug content distributed evenly.
DTPP-TPGS offers greater intracellular drug delivery due to its rapid endocytosis and subsequent endosomal escape. DTPP-TPGS also efficiently
inhibits efflux transporter P glycoprotein (PgP), which, along with greater cell uptake and inherent cytotoxic activity of the construction material
(TPP-TPGS), cumulatively results in 3-fold increment in anticancer activity of DOX in resistant breast cancer cells as well as greater induction of
necroapoptosis and arrest in all phases of the cell cycle. DTPP-TPGS after intravenous administration in Balb/C mice with breast cancer accumulates
preferentially in tumor tissue, which produces significantly greater antitumor activity when compared to DOX solution.
[Molecular Pharmaceutics, 2019, 16, 3744]
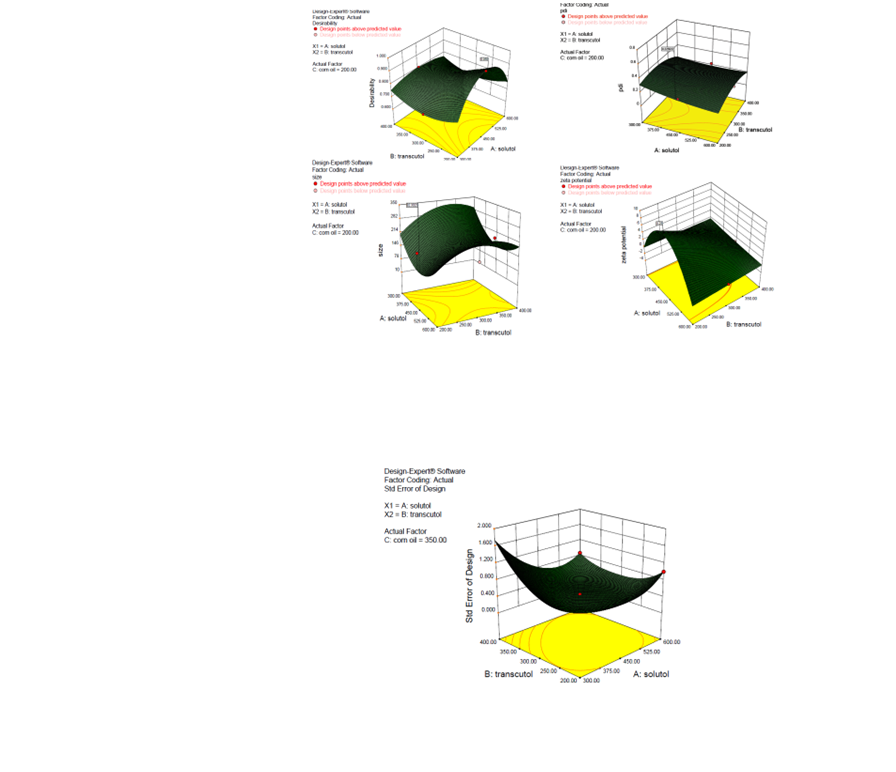
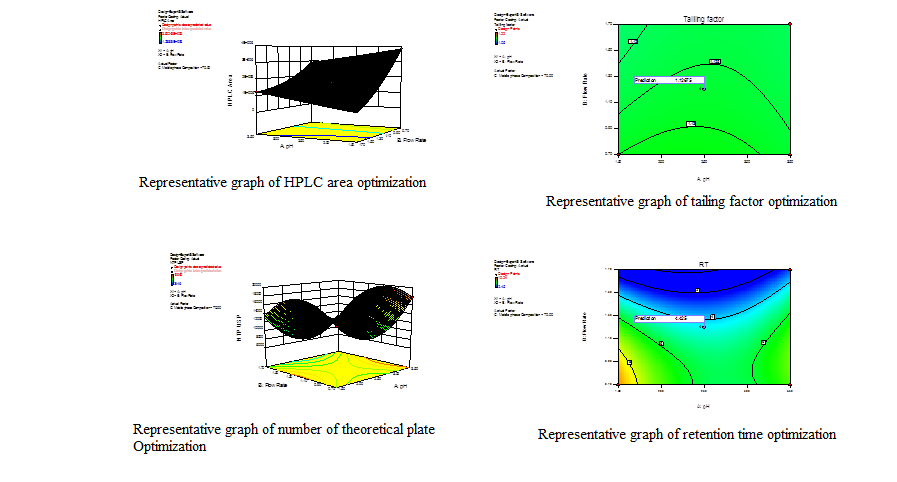
Analytical method development and validation utilizing QBD approach for oral sustained-release formulation of Fenofibrate intended to control alcohol –induced dose dumping
The peformulation and formulation development studies have been performed, to develop oral sustained-release tablet formulation of fenofibrate intended
for controlling alcohol induced dose dumping. Further, a robust analytical method was developed for estimation of fenofibrate deliberated for content
uniformity test, blend uniformity test, multimedia in-vitro studies, and dose dumping studies. The analytical method development has been done
utilizing a novel and rational quality by design approach.
Development of self emulsifying delivery system bearing Moringa oleifera extract
This particular work involves formulation of self emulsifying drug delivery system intended for oral delivery of extract of Moringa oleifera.
The formulation involves selection of surfactant, co-surfactant and oil phase in accordance with solubility study of extract in different vehicles
involving FDA approved oil, surfactant, and co-surfactant. The preformulation studies suggested compatible sets of excipients that can best
accommodate the herbal extract providing a large window for dose adjustment. The compatibility study was conducted in 14 different ratio of oil
and surfactant-cosurfactant mixture. The selected excipients were subjected for various formulation trials to get most favouring formulation on the
basis of particle size, polydispersity index, emulsification volume & time in simulated gastric fluid, simulated intestinal fluid and water.
The formulation was rationally optimised using quality by design approach i.e. involving response surface methodology using Box–Behnken design.
After development, formulations were characterized for various physicochemical and pharmaceutical attributes.