Development of Multi-Component Crystalline Pre-formulation of Drugs:
Physicochemical properties such as solubility, stability (thermal and mechanical) and hygroscopicity are crucial for the translation of active
compounds for drug development. Many active compounds fail to show favourable water solubility and hence results in poor bioavailability.
Solubility is a bulk property which depends on stability of crystalline phase which in turns depends on the internal arrangement of molecules in
crystal. Development of new cocrystals and organic salt forms of the parent compound provide a new approach to alter molecular packing
arrangements in crystal and hence results in the alteration of their physicochemical properties. These molecular packing alterations require
a thorough understanding of intermolecular interaction strength and types. Design, synthesis and characterization of these solid forms can be
used to further expand the scope of the available pre-formulation options beyond pure API forms.
Crystal engineering approaches such as supramolecular synthon complementarity, crystal structure prediction (CSP), experimental charge density (ECD) studies
play crucial role in the design and synthesis of these new crystalline phases. Our group routinely employs these techniques in the rational design of
multicomponent crystals of drugs and pharmaceutically relevant compounds developed within the institute.
References:
• New crystalline salt forms of levofloxacin: Conformational analysis and attempts towards the crystal structure prediction of the anhydrous form. CrystEngComm, 2014, (DOI: 10.1039/c4ce00263f).
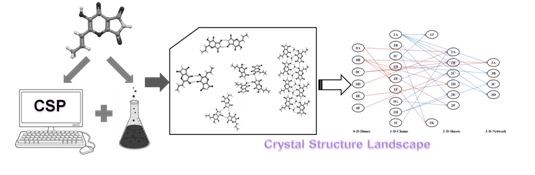
Crystal Structure Prediction (CSP) Based study of Crystal Structure Landscape:
Selection of an optimal crystalline form of drug is crucial for the industrial scale manufacturing, processing and storage of drugs as they are known to
exhibit phase transformation to new polymorphs and solvates forms during processing and storage. An exhaustive screening of various possible crystalline
forms of a drug is very important to the pharmaceutical industry. Crystal structure prediction (CSP) computations plays an important role in obtaining the
structural information for the polymorphs, solvates and other multicomponent crystals in the absence of experimental data. It serves as an important tool
to predict various plausible crystal structures (space group, lattice parameters, and the positional coordinates of atoms) for a given single or
multicomponent system before its experimental realization. The crystal structure landscape constitutes it’s all possible crystalline forms
(polymorphs and pseudopolymorphs) of a compound as these crystal forms represent distinct crystallization pathways. The identification of all of
these crystallization pathways and structures by means of a CSP study can be very useful in sampling the crystal structural landscape computationally.
References:
• Experimental and computational crystal structure landscape study of nigerloxin: A fungal metabolite from aspergillus niger. CrystEngComm2016 (DOI: 10.1039/c5ce02572a).
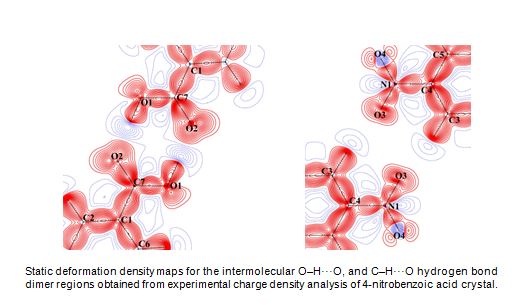
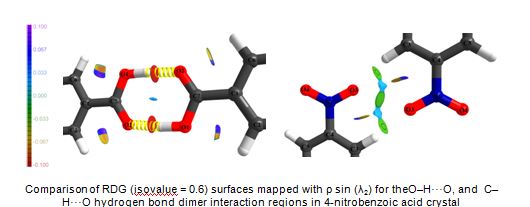
Experimental Charge Density Studies:
A quantitative study of various typesof intermolecular interactions present in crystals such as hydrogen bonds, halogen bond, π...π, dipole...dipole
interactions etc. is essential for the understanding of molecular recognition phenomenon in chemistry and biology. The electron density distributions
obtained from the multipole modeling of high resolution X-ray diffraction data provides an excellent means for studying these intermolecular interactions
experimentally. Our research group works on the experimental X-ray charge density study of various intermolecular interactions in molecular crystals.
The topological analysis of the electron density distribution performed under the framework of Bader’s quantum theory of atoms in molecules (QTAIM) at
the intermolecular regions, molecular electrostatic potential, Hirshfeld surfaces, reduced density gradient (RDG) based descripterand multipole derived
electrostatic interactions energies are used by us to obtain both qualitative and quantitative information on the nature and strength of various
intermolecular interactions. A charge density study requires a very high quality crystal and diffraction data of subatomic resolution (< 0.5 Å).
References:
• Studying the role of C=O...C=O, C=O...N−O, and N−O...N−O dipole−dipole interactions in the crystal packing of 4‑nitrobenzoic acid and 3,3′-dinitrobenzophenone polymorphs: an experimental charge density study. Cryst. Growth Des. 2015, (DOI: 10.1021/acs.cgd.5b00408).